In the toolbox of most scientists over the past century, there have been few methods as powerful and as versatile as linear regression. The introduction of the generalized linear model (GLM) framework in the 1970’s extended the inferential and predictive capabilities to binary or count data. While the effect of this ‘Swiss Army knife’ of scientific research cannot be overstated, rapid (and amazing) technological advances in other areas have pushed it beyond its theoretical capacity.
Linear regression is obtained by applying Gauss’ ordinary least squares (OLS) method, which relies on having a sample size which is sufficiently larger than the number of predictors. However, in this age of ‘high throughput’ data, such as RNA sequencing, the number of predictors (say, the expression level of thousands of genes) greatly exceeds the number of observations. This is often called the ‘large P, small n’ problem. In such cases, the OLS procedure does not work, and the natural next step is to perform variable selection and choose a small number of predictors which are associated with the response variable.
The question is, how to choose the right predictors? A good selection method should detect most (ideally, all) the true predictors, but not at the expense of getting too many false ones (in the ideal case, it would yield none.) This has been one of the greatest challenges in statistics in the past 25 years. The most widely used approach is based on adding a ‘penalty term’ to the optimization problem, so as to practically put a constraint on the number of predictors which can have a non-zero effect on the outcome. Among the methods which rely on this approach, the most well-known is the lasso. The approach we take in our paper is different. We assume that the effect predictors have on the outcome can be either positive, negative, or have no effect at all (which we call null predictors). Mathematically, we model it as a three-component mixture in which the effects of the non-null predictors are assumed to follow normal distributions. This approach is related to the Bayesian ‘spike-and-slab’ approach. The difference is that in the spike-and-slab model, the non-null predictors are assumed to follow a single normal distribution with mean 0. The difference between the two approaches is illustrated in Figure 1.

Figure 1 A graphical representation of the spike and slab model vs. our mixture model
Real data, however, is noisy, which means that the measured effects of the null predictors is not a spike at zero. Rather, it is a Gaussian distribution which has a fair amount of overlap with the distributions of the true predictors. This is what makes the variable selection task so hard, regardless of which approach is used.
The three-component mixture of normal distributions which we use in our model allows us, with a simple adaptation, to perform variable selection in the GLM framework. For example, we apply it to binary-type response in order to identify which gut bacteria is associated with obesity status. It can also be used to perform variable selection in survival analysis by modeling the number of new events at time t as a Poisson process. We identify five genes which are associated with survival probability of breast cancer patients.
Our method (which is implemented in an R package called SEMMS) includes three important features. First, it allows the user to ‘lock in’ predictors, which means that it is possible to choose certain substantively important variables to always be included in the model, and not be subject to the selection procedure. Second, for the initialization of the algorithm we can use the variables selected by other methods (e.g., lasso, MCP) and because the algorithm will never result in a decrease in the log-likelihood from one iteration to the next, our final model will always be at least as good as the one obtained by the other variable selection method which was used in the initialization step. Third, the model accounts for correlations between pairs of predictors. This greatly alleviates the problem of identifying true predictors in the presence of multicollinearity. Methods based on penalization often struggle in the presence of multicollinearity. This is demonstrated in the following example, where the objective is to identify which of 4,088 genes is associated with the production rate of riboflavin. Out of 8,353,828 possible pairs of genes, 70,349 have correlation coefficient greater than 0.8 (in absolute value). This high degree of multicollinearity, combined with the relatively small sample size (N=71) makes this dataset especially challenging for variable selection methods. The model selected by SEMMS results in much smaller residuals than ones obtained from other variable selection approaches. It also provides much better prediction for low values of riboflavin.
We conclude with a few words of caution. No matter which method is used, beware of declaring the selected model as the “best” because (i) the number of putative variables is so large that there is no way to evaluate all possible models, and (ii) some selected predictors can be part of a network of highly correlated variables. The riboflavin data provides an excellent example, which is illustrated in network diagram in Figure 2.
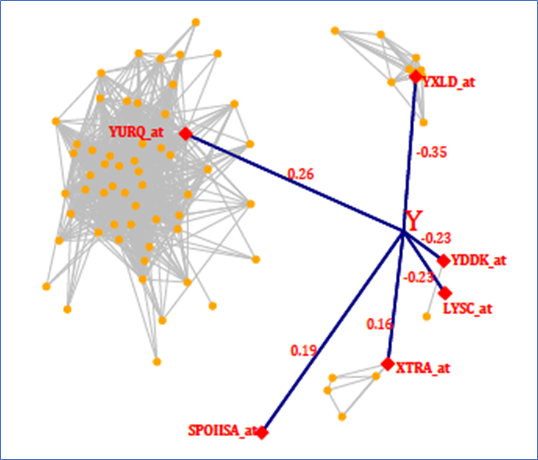
Figure 2 Riboflavin data – a graphical representation of the model found by SEMMS
SEMMS detects six strong predictors for riboflavin production rate (shown as red diamonds), but it also detects that four of them are highly correlated with other genes (orange dots). One gene (YURQ_at) is co-expressed with a large group of genes, each of which could be considered as a relevant predictor for the response. A network diagram like this helps to illustrate not only that there may be no single “best” model, but also that there may be complicated interactions between the predictors, and thus, the relationship between the predictors and the response may not be linear.
 Haim Bar is an Associate Professor in Statistics at the University of Connecticut. His professional interests include statistical modeling, shrinkage estimation, high throughput applications in genomics, Bayesian statistics, variable selection, and machine learning.
Haim Bar is an Associate Professor in Statistics at the University of Connecticut. His professional interests include statistical modeling, shrinkage estimation, high throughput applications in genomics, Bayesian statistics, variable selection, and machine learning.
 James Booth is a Professor in the Department of Statistics and Data Science at Cornell University. He completed his PhD at the University of Kentucky in 1987 working with advisers Joseph Gani and Richard Kryscio, after which he was hired as an Assistant Professor at the University of Florida. He was hired at Cornell in 2004.
James Booth is a Professor in the Department of Statistics and Data Science at Cornell University. He completed his PhD at the University of Kentucky in 1987 working with advisers Joseph Gani and Richard Kryscio, after which he was hired as an Assistant Professor at the University of Florida. He was hired at Cornell in 2004.
 Martin T. Wells is the Charles A. Alexander Professor of Statistical Science and Chair of the Department of Statistics and Science at Cornell University. He also has joint appointments in the Cornell Law and Weill Medical Schools.
Martin T. Wells is the Charles A. Alexander Professor of Statistical Science and Chair of the Department of Statistics and Science at Cornell University. He also has joint appointments in the Cornell Law and Weill Medical Schools.